 The Medical Device Regulation (MDR) aims to harmonize the European market for medical devices.
The Medical Device Regulation (MDR) aims to harmonize the European market for medical devices.
This blog series discusses the Medical Device Regulation (MDR) in brief and how it affects software development for medical devices. In this (last) part we will discuss the general safety and performance requirements and the software lifecycle processes for the medical device software.
Checklist for Medical Software Development (4/4)
The key points to consider regarding the the general safety and performance requirements and the software lifecycle processes:
- Define requirements for information security and cyber security
- Consider the specific features of the mobile platform in the environment the software is used
- Ensure the interoperability and compatibility with other devices or products
- Define requirements and considerations for devices intended for use by lay persons
- Establish, document and maintain system for risk management according to ISO 14971
- Control risks for functionality, usability, performance and security
- Make software safety classification according to IEC 62304
- Define software development process based on the software safety classification
- Define and implement design reviews or controls
- Define and implement an efficient CAPA process
- Ensure traceability through the whole software development lifecycle
- Acquire efficient toolsets
General Safety and Performance Requirements
The Safety and Performance Requirements in MDR Annex I replace the directives’ Essential Requirements. A manufacturer needs to demonstrate compliance with these requirements through risk management, testing, technical studies, and other means. The most important changes in MDR affecting software development include the following:
- Risk management
- Electronic programmable systems and software
- Design for compatibility
- Devices intended for use by lay persons
Risk management addressed in MDR Annex I is essentially a summary of the requirements of ISO 14971. Risk management is also emphasized in ISO 13485:2016.
For software, principles of information security and cyber security are new areas of emphasis in the MDR. Manufacturers shall set out minimum requirements concerning hardware, IT network characteristics and security measures, including protection against unauthorized access. Manufacturers are also required to consider the specific features of the mobile platform (e.g. size and contrast ratio of the screen) and the external factors related to their use (varying environment with regard to the level of light or noise).
Devices that are intended to be operated together with other devices or products need to be designed and manufactured in such a way that the devices are reliable and safe in terms of interoperability and compatibility.
Devices for use by lay persons need to be designed and manufactured in such a way that they perform appropriately for their intended purpose considering the skills and means available to lay persons. The information and instructions (e.g. IFU and labeling) must be easy for lay persons to understand and apply.
Risk Management
Risk management means the systematic application of management policies, procedures and practices to the tasks of analyzing, evaluating, controlling and monitoring risks. Risk management must be performed during the entire medical product life-cycle including the post-production phase. Software risk management must be connected to the risk management of the whole medical device or system.
Risk management process
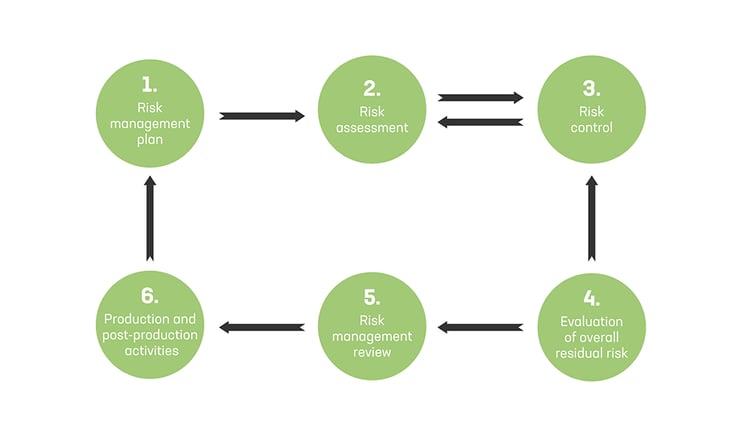
ISO 14971 defines the process steps of the risk management.
ISO 14971 defines the following process steps for the risk management to medical devices:
- Risk management plan (activities, responsibilities, criteria for risk acceptability, verification activities etc.)
- Risk assessment (risk analysis and evaluation)
- Risk control (including option analysis, implementation, residual risk evaluation, risk/benefit analysis, assessment of possible new risks introduced, review of risk control completeness)
- Evaluation of overall residual risk
- Risk management review (of the execution of the risk management plan)
- Production and post-production activities (with feedback to risk management plan and risk assessment)
Scope
Risk management should be implemented at least for:
- Developed and maintained (software) products
- Security (information security and cyber security)
- Risk based approach to QMS processes
- Project risks, financial risks or risks to business performance
Product risk management analyzes, evaluates and controls the product related risks, e.g. for functionality, usability and performance. This may be integrated for example to design reviews or controls.
Security risk management concerns the risk analysis, evaluation and control principles regarding security threats that could impact the confidentiality, integrity or availability of a medical device (including both the software product and the IT operating environment) or information processed by the device with possible impact also to patient safety. Security management is executed throughout the product’s lifecycle through following activities: identification, protection, detection, response, and recovery.
Risk based approach to the control of appropriate QMS processes establishes a basis for maintaining and improving the suitability, adequacy and effectiveness of the QMS. Focus and priority should be on the processes, practices and software that have the highest impact on meeting safety or performance requirements of the medical device or applicable regulatory requirements.
ISO 14971 does not address the business risks. However, project risks, financial risks or risks to business performance must be managed through the business planning.
Traceability between risks, their controls, and testing to verify those controls must be available.
Software Development Lifecycle
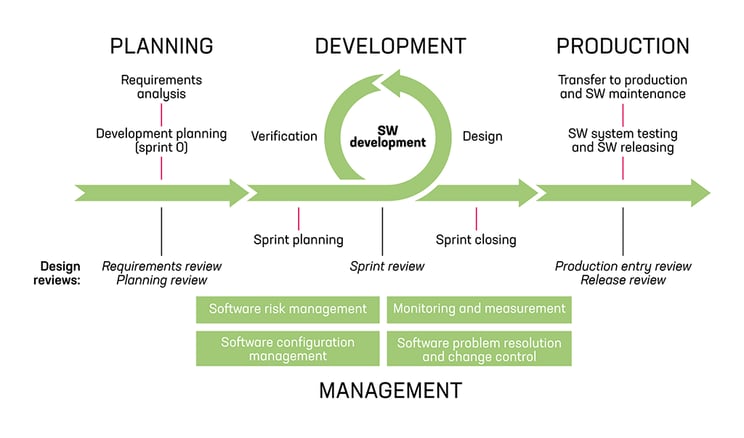
An example of the software development process structure.
The cornerstone of the medical software development process is the software development and maintenance life cycle processes defined in IEC 62304, supported by ISO 14971 for risk management and ISO 13485, e.g. for customer-related processes, as well as design and development processes. Agile practices, like the Scrum, can be integrated to the core of the development cycles.
Software maintenance repeats all the relevant processes used in the initial development project, either as is or separately defined for the maintenance project. The practices to remove the software device or service from the market should be considered as early as it is relevant.
Software safety classification
The core of the IEC 62304 implementation is the software safety classification (classes A, B and C). Patient safety is critical, and ensuring it via testing is not enough. The software safety classification determines the safety-related processes that must be applied for the entire development lifecycle, from requirements and design to release and maintenance. Building safety into processes early on will save time, effort and expenses later.
The software safety class is determined by the evaluation of the risks related to potential software system failures and the severity of injuries possible. The software safety classification may be lowered by identifying and implementing external risk mitigations, e.g. via hardware or other software, so that software failures do not result in an unacceptable risk.
Note that the software safety classification is different to medical device risk classification, and they are both used for different purposes. The first is used to determine the required software processes and the latter is used for medical device regulatory purposes.
Design reviews
Design reviews, together with risk management, ensure that the product is safe and effective for its intended purpose.
Design reviews are moments in time to evaluate the progress of the project and the risks of the product as it progresses through development. Design reviews may be aided with checklists for the relevant steps. For example, the transfer to production is a step where software is placed on the market as a device or put into service in its own right. At the same time, the responsibility of software is transferred from a development project to a maintenance project. This transfer should be gated through a related design review.
CAPA
The Corrective and Preventive Action (CAPA) process is to identify, prevent and eliminate the causes of actual or potential nonconformity related to medical software products and to the Quality Management System, using risk management principles. CAPA is a critical QMS process as several other processes connect with it.
Typical challenges regarding the CAPA process include:
- CAPA is almost always cross-functional in nature and involves many groups and functions
- Most CAPA effort is typically placed on correcting issues rather than preventing them in the first place
- CAPA is overused or underused; it should be reserved as a process to deal with potential systemic issues
- The problem statement and issue description are hardly ever the true root cause
- Poor definition of CAPA process
Data and traceability
Having enough information and reliable data is essential in the development and post-market phases, e.g. for risk management.
For example, product-related customer complaints or non-conformances need to be traced back to specific design control elements.
Another vital aspect is the traceability, not only to trace requirements to implementation and testing, but to connect people, processes and data seamlessly across the value chain. For example, product-related customer complaints or non-conformances need to be traced back to specific design control elements. CAPA ties the data from the PMS processes back to design and development through traceability and risk management.
In addition, traceability should be bidirectional, e.g. from requirements to test cases, and from test cases back to requirements. Traceability is also checked in the audits by your Notified Body.
Tools
Tools play a vital role in efficient software development processes. One is the toolset for version control, configuration management, continuous integration and continuous delivery. In addition, tools for requirements management, agile practices and risk management are a must. Having a complete product lifecycle management toolset with a regulatory setup is worth considering.
It may be challenging to provide full ISO 13485 or MDR compliancy via plain papers or documents or spreadsheet solutions in the software development process. Proper use of tools also enables traceability, e.g. for change management. Tools and software applications used in the software development lifecycle need to be validated prior to their initial use, and after changes as appropriate, based on the risk associated with the use.
Download our free guide: Medical Device Regulation on Software Development – Key points to consider
Pinja's quality management system for health and welfare technology is ISO 13485:2016 certified.

References:
ISO 13485:2016
ISO 14971:2019
IEC 62304
Medical Device Regulation (EU) 2017/745
Guidance on Cybersecurity for medical devices (MDCG 2019-16)
Pinja Welfare and Health Technology, Quality Management System (internal document)
Read more
Medical Software Development according to Medical Device Regulation (MDR) – Part 1: Software Qualification and Classification
Medical Software Development according to Medical Device Regulation (MDR) – Part 2: Conformity assessment route, software modules and impact of changes
Medical Software Development according to Medical Device Regulation (MDR) – Part 3: Quality Management System
Guide: Medical Device Regulation and Software Development – Key points to consider
Synoste success story
BioMediTech success story
Health and welfare technology
