This blog series discusses the Medical Device Regulation (MDR) in brief and how it affects software development for medical devices. In this first part we will look at the MDR in brief and the software qualification and risk classification.
MDR in Brief
The ever-changing regulations and standards and the higher importance of software in devices and systems used in healthcare has made medical software development very challenging. The Medical Device Regulation (EU) 2017/745 (MDR) aims to harmonize the European market for medical devices, building on the directives for medical devices and active implantable medical devices (MDD/AIMDD).
MDR changes the scope of the responsibilities that medical device manufacturers must control. First, they must control the whole supply and distribution chain. MDR introduces the term economic operators, covering not only the manufacturers but, e.g. distributors and importers as well. For example, the manufacturer is responsible for the training of all its economic operators.
Manufacturers must control the whole lifetime of a medical device.
Secondly, MDR promotes a shift from pre-market approval (i.e. the path to CE marking) towards an entire lifecycle approach. Therefore manufacturers must control the whole lifetime of a medical device from early clinical evaluations and investigations through design, development and placing the device on the market, or putting it into service, to post-market surveillance and clinical follow-up of the devices or service (including product and process changes), and finally taking the device off the market or out of service.
The list of device categories to fall under the scope of the MDR has become broader. At the same time the medical device risk classification has been changed with the impact that most software products may be classified into class IIa as a minimum. All this means that manufacturers will need to assess their product portfolio and find where there may be an impact due to the regulatory changes.
The MDR requires a manufacturer to have a Quality Management System (QMS) in place. The big adjustment with the MDR updates is the shift from items being directives (MDD/AIMDD) to becoming regulations (i.e. laws) in the EU.
Important Dates
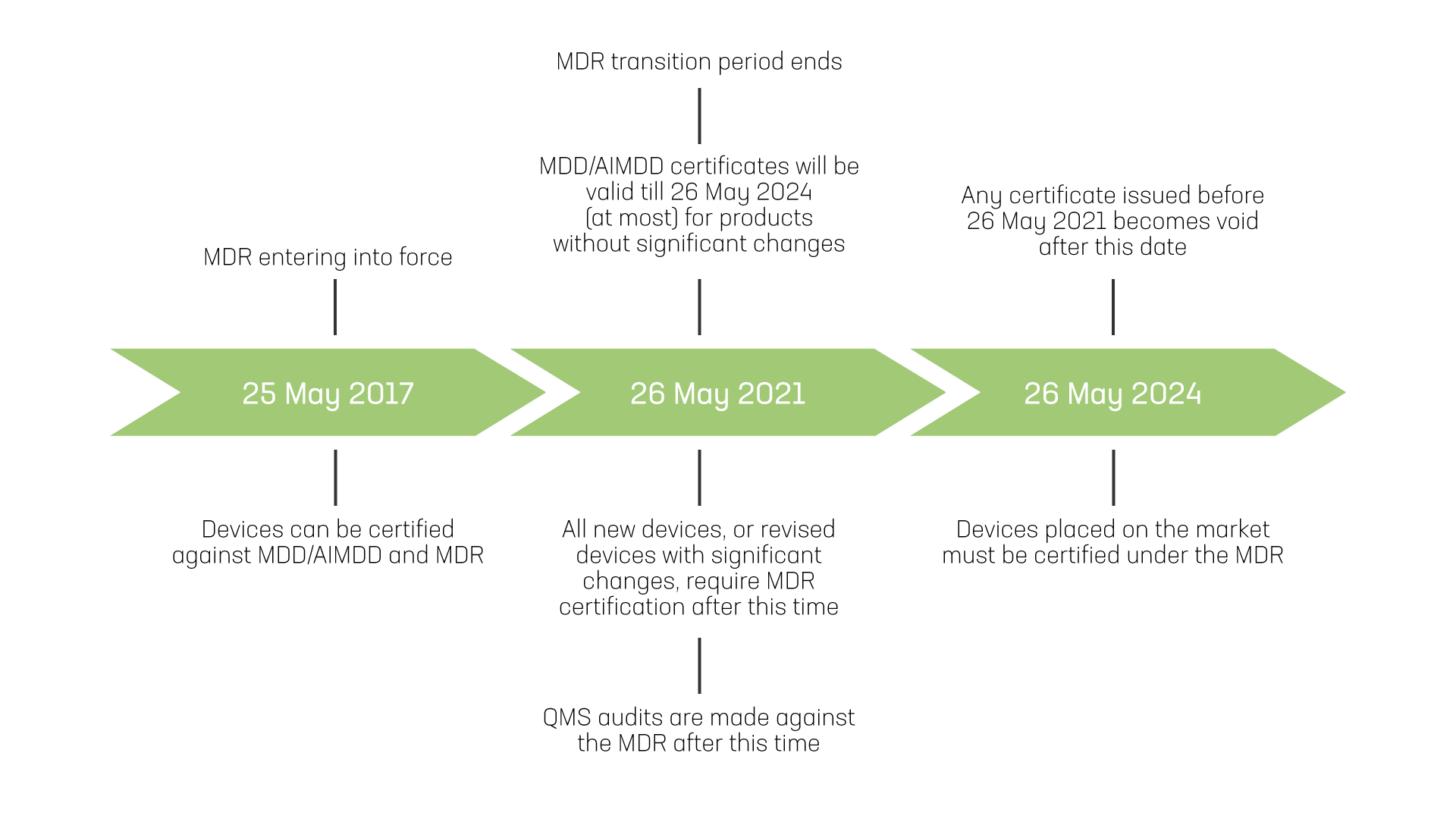
Important dates in MDR implementation for medical device. Guidance for identifying significant changes is discussed in the later blogs.
Checklist for Medical Software Development (1/4)
The key points to consider regarding the software qualification and risk classification:
- Review the device/software and its intended purpose for medical device qualification
- Perform risk analysis for impacts on using the device/software for its intended purpose
- Determine the risk classification for the medical device/software
- Start Notified Body intervention (for risk classes other than class I self-certified)
Qualification of Software as a Medical Device
For the purposes of the MDR, a medical device means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific medical purposes defined in MDR Article 2.
MDR addresses the higher importance of the software in the devices and systems, as well as the introduction of software as medical devices. Software may be, e.g. embedded software in a device, a mobile application, a web service or an expert system or a component in such a system. If a device is qualified as a medical device, it will be covered by the MDR and shall be certified accordingly.
Software qualification guidance
The European Commission has released the Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745. MDR is to be applied basically for two types of software:
- Medical device software (MDSW)
- Software driving or influencing the use of a medical device
Medical device software (MDSW)
Software must have a medical purpose on its own to be qualified as MDSW. MDSW is software that is intended to be used, alone or in combination, for a medical purpose as specified in the MDR, regardless of whether the software is independent, driving or influencing the use of a device.
Software may be qualified as MDSW regardless of its location (e.g. operating in the cloud, on a computer, on a mobile phone, or as additional functionality on a hardware medical device). MDSW may be intended to be used by healthcare professionals or lay persons (e.g. patients or other users).
 Software may also be intended to drive or influence the use of a (hardware) medical device.
Software may also be intended to drive or influence the use of a (hardware) medical device.
Software driving or influencing the use of a medical device
Software may also be intended to drive or influence the use of a (hardware) medical device, but does not have to perform a medical purpose on its own, or it does not create information on its own for one or more of the medical purposes of a medical device. This kind of software that is driving or influencing the use of a medical device is covered by the MDR either as a part or component of a device or as an accessory for a medical device.
Non-medical device software
The guidance also highlights the questions; is the software performing an action on data different from storage, archival, communication or simple search, or is the action for the benefit of a patient group or larger population instead of individual patients. Therefore, e.g. general patient health records software or an appointment booking application is not a medical device, while a monitoring or analysis application may be a medical device.
Similarly, e.g. a mobile application for managing pictures of moles is not a medical device while an application for the assessment of moles may be a medical device. The guidance document includes further instructions and examples across the different qualification decision steps.
Intended purpose
It should be noted that the intended purpose of the device or software, as described by the manufacturer, is relevant both for the qualification and classification of any device. In addition, the broader device scope of the MDR may change the device becoming a medical device in its intended purpose.
The analysis for intended purpose should take into account – for example – the following:
- What is the target patient or age group
- Who uses the device, and are there needs for specific competence
- Who uses the generated data and for the benefit of whom
- What is the environment of use
- What vital functions will be affected directly or indirectly
- In what phase of treatment the device is used (preliminary, operation or monitoring)
- How device derives the input into output (produce, transfer, change, measure, screen, support, etc.)
- Does the device change or replace some current function or does it provide a completely new one
Risk Classification of a Medical Device and Software
Determining the risk class of a medical device is essential in specifying the required steps for CE marking and certain post-production activities.
Medical devices shall be divided into classes I, IIa, IIb and III, considering the intended purpose of the devices and their inherent risks. Determining the risk class of a medical device is essential in specifying the required steps for CE marking and certain post-production activities.
The risk analysis must consider both the intended (correct) use and use errors as well as abnormal use. Risk analysis helps in determining the risk class of the device and guide in defining or refining the intended purpose (e.g. target patient group or user competence).
Classification rules
For the classification itself all implementing rules in MDR Annex VIII shall be considered. Rule 11 defines the class IIa as a ‘default’ software classification:
- Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa
Rule 11 further details this, together with rules 15 and 22:
- Class III if decisions have an impact that may cause death or irreversible deterioration of a person’s state of health
- Class III for active therapeutic devices with an integrated or incorporated diagnostic function, which significantly determines the patient’s use of the device, such as closed loop systems or automated external defibrillators
- Class IIb if decisions have an impact that may cause serious deterioration of a person’s state of health or a surgical intervention
- Class IIb if software is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient
- Class IIb if software is used for contraception
- Class I for all other software
These rules basically define most software at least to class IIa. There are additional requirements in rules 3.3 and 3.5:
- Software, which drives or influences the use of a device, shall fall within the same class as the device
- If software is independent of any other device, it shall be classified in its own right
- Software that both achieves its own intended purpose and also drives or influences the use of a (hardware) device for a medical purpose is classified on its own, but the risk class shall not be lower than the risk class of the hardware medical device
Classification summary
The Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 includes further instructions and examples for defining the device risk class. The guidance document illustrates a matrix of significance of information and criticality of the situation or condition (excluding the class I):

The matrix of significance of information and criticality of the situation or condition.
Classification impacts
The classification of the device or software has the following impacts for the manufacturer:
- QMS must be defined and implemented; for classes other than class I (self-certified) the QMS must be in accordance with the MDR
- Notified Body intervention is required for classes other than class I (self-certified)
Download our free guide: Medical Device Regulation on Software Development – Key points to consider
References:
Medical Device Regulation (EU) 2017/745
Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745
Read more
Blog: Medical software development according to Medical Device Regulation (MDR) – part 2: Conformity assessment route, software modules and impact of changes
Blog: Medical Software Development according to Medical Device Regulation (MDR) – part 3: Quality management system
Blog: Medical software development according to Medical Device Regulation (MDR) – part 4: Medical device software lifecycle processes
Guide: Medical Device Regulation and Software Development – Key points to consider
Evondos success story
Synoste success story
BioMediTech success story
Welfare and health technology
